



La farmacocinética es la rama de la farmacología relacionada con la descripción matemática del curso temporal de las concentraciones plasmáticas del fármaco medidas después de la administración de una dosis. Específicamente, la farmacocinética es el uso de modelos matemáticos para describir cómo se comporta un fármaco en el organismo durante la absorción, la distribución, el metabolismo y la excreción (procesos conocidos en conjunto como ADME). En términos simples, la farmacocinética describe lo que el organismo le hace al fármaco, mientras que la farmacodinámica describe lo que el fármaco le hace al organismo.
Los parámetros farmacocinéticos se calculan siguiendo los cambios en los datos de concentración plasmática frente al tiempo del fármaco después de que se administre una dosis del fármaco, al menos por la vía deseada, e idealmente también después de la administración IV (100 % de biodisponibilidad). Los modelos matemáticos desarrollados deben permitir predicciones sobre el movimiento del fármaco en el organismo y su eliminación, de modo que se puedan diseñar pautas de dosificación óptimas y, cuando sea necesario, se puedan estimar los tiempos de retirada del fármaco (para competición, seguridad alimentaria).
La mayoría de los estudios farmacocinéticos se llevan a cabo en animales sanos, pero los protocolos de dosificación deben individualizarse para ajustarse a las diferencias en la fisiología (edad, sexo, especie y raza), la farmacología (interacciones farmacológicas) y la patología (p. ej., enfermedad renal o hepática).
Lo ideal es que las predicciones realizadas utilizando modelos farmacocinéticos se validen en pacientes individuales mediante la recogida de muestras para la monitorización terapéutica de los fármacos. Esta validación es especialmente importante para fármacos con un índice terapéutico estrecho, es decir, un rango limitado de concentraciones tisulares eficaces en las que las concentraciones demasiado bajas son ineficaces o las concentraciones demasiado altas son tóxicas (p. ej., la gentamicina).
La respuesta farmacodinámica a un fármaco suele reflejar el número de receptores con los que interactúa el fármaco (teoría fármaco-receptor). En la mayoría de los casos, las concentraciones tisulares del fármaco son paralelas a las concentraciones plasmáticas del fármaco, razón por la cual los datos plasmáticos pueden utilizarse por lo general como sustitutos de las concentraciones tisulares.
Los parámetros farmacocinéticos más relevantes que afectan al movimiento de un fármaco y proporcionan la base para la pauta posológica son el volumen de distribución aparente (Vd) y el aclaramiento plasmático (Cl), los cuales determinan la tasa de eliminación constante (kel) y la semivida de eliminación (t½). Los parámetros adicionales pueden incluir la tasa de distribución constante y la tasa de semivida y, si el fármaco se administra PO, la tasa de absorción constante y la semivida de absorción.
Después de administrar un fármaco mediante una inyección IV rápida (p. ej., en bolo), el fármaco se distribuirá inmediatamente a través del compartimento vascular central, que incluye órganos muy perfundidos. También, inmediatamente, las concentraciones plasmáticas del fármaco disminuyen debido a que la distribución del fármaco desde el plasma a los tejidos es más rápida que el retorno del fármaco desde los tejidos al plasma, y a la eliminación del organismo (es decir, eliminación irreversible) a través del metabolismo y la excreción. Por tanto, la disminución de las concentraciones plasmáticas del fármaco inicialmente es rápida; sin embargo, una vez que la distribución alcanza un pseudoequilibrio tal que la cantidad de fármaco que se mueve hacia los tejidos es igual a la que regresa al plasma, la concentración plasmática del fármaco disminuirá solo debido a la eliminación del organismo (metabolismo y excreción).
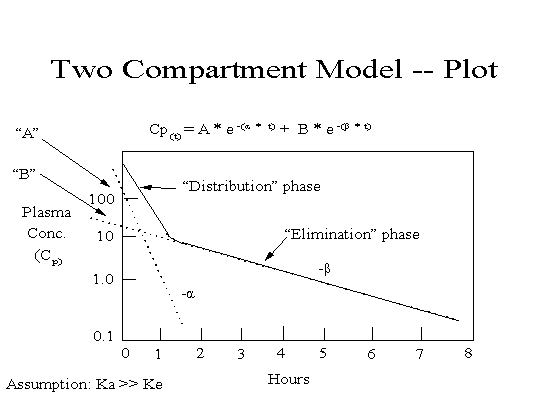
Con cada movimiento del fármaco, suele moverse una fracción o porcentaje constante (en lugar de una cantidad) por unidad de tiempo (es decir, cinética de primer orden). Suponiendo una farmacocinética lineal (proporcionalidad directa entre la dosis y la exposición), las concentraciones plasmáticas del fármaco representadas en una escala semilogarítmica (es decir, una escala logarítmica en el eje y y una escala lineal en el eje x) pueden ajustarse mediante una línea recta (lo que sugiere que los datos están descritos por un modelo farmacocinético de un compartimento) o una curva bifásica (los datos están descritos por un modelo de dos compartimentos).
En un modelo de dos compartimentos, la primera porción de la curva de concentración frente al tiempo (fase de distribución), con una pendiente más pronunciada, representa la distribución y la eliminación. La porción terminal de la curva (fase de eliminación), con una pendiente más plana, representa principalmente la eliminación (es decir, la distribución del fármaco dentro y fuera de los tejidos está en equilibrio, y la distribución en los tejidos ya no contribuye de forma apreciable a la disminución de las concentraciones plasmáticas).
Las constantes de velocidad macroscópicas se pueden determinar para las fases de distribución y eliminación mediante la extrapolación al eje y en un proceso llamado "eliminación de curvas" (consulte el modelo bicompartimental). La intersección con el eje y de la línea que describe la fase de distribución se designa A, y la intersección con el eje y de la línea que describe la fase de eliminación se designa B.
El valor absoluto de la pendiente de la fase de eliminación es la constante de velocidad de eliminación (a menudo denominada beta o kel), y de ella se deriva la semivida de eliminación (t1/2).
El valor absoluto de la pendiente de la recta que describe la fase de distribución es la llamada tasa de distribución constante (a menudo denominada alfa o kd). Esta constante de tasa permite el cálculo de distribución de la semivida.
Una vez que los datos se describen matemáticamente por las pendientes y las intersecciones y de estas líneas, la concentración plasmática del fármaco en cualquier momento dado después de la administración del fármaco puede predecirse mediante la siguiente ecuación:


donde Cp(t) es la concentración plasmática en función del tiempo, A es la intersección con el eje y de la línea que describe la fase de distribución, α es la tasa constante para la porción de distribución de la curva de concentración plasmática frente al tiempo, B es la intersección con el eje y de la línea que describe la fase de eliminación, β es la tasa constante para la porción de eliminación de la curva de concentración plasmática frente al tiempo, e es la base del logaritmo natural y t es el tiempo transcurrido desde la administración.
A partir de estos modelos, los parámetros clínicamente relevantes (Vd y Cl) se pueden calcular y utilizar para determinar las pautas posológicas adecuadas.
Volumen de distribución aparente
El parámetro farmacocinético utilizado para evaluar la extensión de la distribución del fármaco por todo el organismo se conoce como volumen de distribución aparente (Vd). Si se conocen tanto la dosis como la concentración plasmática del fármaco, el Vd se puede calcular de la siguiente manera:

donde Vd es el volumen de distribución aparente (en L/kg), D es la dosis (en mg/kg) y Cp es la concentración plasmática del fármaco (en mg/l).
Este volumen teórico es el volumen al que debe distribuirse el fármaco si la concentración en plasma representa la concentración en todo el organismo. El término "aparente" subraya el hecho de que el Vd no indica dónde se distribuye el fármaco a partir del volumen de distribución, sino que solo va a alguna parte.
Hay varias formas de calcular el Vd, según el fármaco y la necesidad clínica. Para los agentes anestésicos inyectables, la concentración en el compartimento central o vascular (que incluye el cerebro y los órganos altamente perfundidos) es la más importante, porque esta concentración refleja muy probablemente la concentración del agente anestésico en el cerebro. En este caso, puede estimarse el Vd del compartimento central (Vdc):

Para la mayoría de los demás fármacos, el Vd en estado estacionario (Vdss), que refleja el volumen que parece estar ocupado cuando la distribución tisular alcanza el pseudoequilibrio, se calcula sumando el Vdc y el volumen teórico del segundo compartimento tisular (V2). V2 se calcula a partir de las constantes de velocidad estimadas por software de modelado matemático durante el proceso de ajuste de datos.
El Vd es útil por varias razones. Quizás la más importante es que puede usarse para calcular una dosis si se conoce la Cp objetivo, mediante reordenamiento de la ecuación para el Vd. Por ejemplo, si el Vd en estado estacionario (Vdss) de fenobarbital es de 0,6 L/kg y la concentración objetivo de fenobarbital en un animal que nunca ha recibido el fármaco es de 10 mg/l, la dosis IV se calcula de la siguiente manera:

Esto es especialmente útil para calcular las dosis de carga de los agentes anestésicos; sin embargo, debe usarse el Vdc para ese propósito.
Una razón adicional por la que el Vd es útil es que si se conoce la Cp en cualquier momento después de administrar la dosis, entonces el Vdss se puede utilizar para calcular cuánto fármaco queda en el organismo.
Finalmente, puede usarse el Vdss para predecir la capacidad relativa del fármaco para moverse a diferentes compartimentos del organismo. Si se limita al compartimento extracelular (líquido intersticial, plasma), como es típico de los fármacos hidrosolubles, el fármaco representa el 20-30 % del peso corporal, y el Vd de dicho fármaco debe ser <0,3 L/kg.
Los fármacos liposolubles suelen ser capaces de atravesar las membranas celulares y, por lo tanto, se distribuyen tanto en el líquido extracelular como en el intracelular, lo que representa ~60 % del peso corporal. Estos fármacos se suelen caracterizar por un Vd >0,6 L/kg. Algunos fármacos se limitan al compartimento plasmático y no se distribuyen bien. Por ejemplo, para fármacos muy ligados a las proteínas plasmáticas, el Vd se aproxima al volumen del compartimento sanguíneo, o <0,1 L/kg. A medida que el fármaco se libera de las proteínas, sin embargo, dejará el compartimento plasmático y se distribuirá a los tejidos.
Muchos fármacos se caracterizan por un Vd que excede el peso corporal del animal (es decir, >1 L/kg). Por ejemplo, la media del Vd de la digoxina en perros es de 13 L/kg. Esto significa que la digoxina se une apreciablemente a otros tejidos (es decir, si el fármaco abandona el plasma, independientemente de dónde vaya, el Vd aumentará).
El Vdss de un fármaco suele ser constante en un amplio rango de dosis para una especie determinada, y es la mejor estimación del Vd para extrapolar entre especies. Sin embargo, una serie de factores clínicamente significativos pueden influir en el Vd, incluyendo la edad (el Vd es mayor en neonatos y pediatría, menor en geriatría); el estado funcional de los riñones (el Vd disminuye con la deshidratación), el hígado (el Vd aumenta con un edema) y el corazón; las acumulaciones de líquido; la concentración de proteínas plasmáticas (influyendo solamente en el fármaco libre); el estado ácido-base (especialmente si el atrapamiento de iones hace que el fármaco se acumule en los tejidos); los procesos inflamatorios o la necrosis (tiende a aumentar la distribución); y cualquier otra causa de alteración en la extensión de unión a proteínas plasmáticas.
Eliminación de los fármacos
Tan pronto como un fármaco alcanza la circulación sistémica, inmediatamente comienza a eliminarse del plasma. El aclaramiento es el volumen de sangre desde la cual un fármaco se elimina o aclara irreversiblemente. Por lo general se muestrea el plasma; sin embargo, el aclaramiento plasmático representa la suma de los aclaramientos de todos los órganos. Si el fármaco es eliminado por un solo órgano, entonces el aclaramiento plasmático es el aclaramiento de ese órgano.
Una definición alternativa de aclaramiento es el volumen de plasma que contendría la cantidad de fármaco excretado por unidad de tiempo. Esta definición demuestra el vínculo entre el volumen de distribución (Vd) y el aclaramiento. Si se conoce la tasa de eliminación constante (kel), esta describe la fracción de Vdss aclarado, y juntos, estos dos valores se pueden usar para calcular el aclaramiento:

donde Cl es el aclaramiento (en ml/kg/minuto), Vdss es el volumen de distribución aparente en estado estacionario (en ml/kg) y kel es la tasa de eliminación constante (en minutos-1).
Como tal, al igual que el Vd, el Cl influye directamente en la kel, es decir, la tasa a la que el fármaco se elimina del organismo: a medida que aumenta el Cl, la kel se vuelve más pronunciada. El aclaramiento es independiente del Vd de un fármaco y, por tanto, de la concentración del fármaco en la sangre; no importa cuánto fármaco haya en la sangre, se eliminará el mismo volumen por unidad de tiempo.
Los dos órganos principales responsables del aclaramiento son el hígado y los riñones. Después de que un fármaco se metaboliza, se elimina irreversiblemente del organismo. Los metabolitos se excretan principalmente a través de los riñones.
El aclaramiento hepático se define como el volumen plasmático totalmente aclarado por unidad de tiempo a medida que la sangre pasa por el hígado. La tasa de depuración hepática depende de la administración del fármaco al hígado, es decir, el flujo sanguíneo (Q) y la proporción de extracción (E) del fármaco, o fracción del fármaco extraído a medida que pasa por el hígado. La extracción, a su vez, está determinada por el aclaramiento intrínseco (capacidad metabólica) del hígado.
Los fármacos eliminados por el hígado se dividen en dos categorías principales. Los fármacos de flujo limitado se extraen tan rápidamente que Q se convierte en el factor limitante del aclaramiento hepático. La unión a proteínas plasmáticas no influirá en el aclaramiento de estos fármacos.
Por el contrario, el paso que limita la velocidad de los fármacos de capacidad limitada es el aclaramiento intrínseco, la capacidad metabólica del hígado. Para estos fármacos, la unión a las proteínas séricas disminuirá la tasa de eliminación. Por tanto, los fármacos altamente unidos a proteínas se denominan "de capacidad limitada, sensibles a la unión", en contraposición a los fármacos que no se unen demasiado a proteínas y, por tanto, de "capacidad limitada, insensibles a la unión".
La enfermedad hepática afecta diferencialmente a los fármacos de flujo y de capacidad limitada. El aclaramiento hepático de los fármacos de flujo limitado disminuye notablemente con cambios en el flujo sanguíneo hepático, como podría ocurrir con la derivación portosistémica. Cuando se administran PO, estos fármacos se suelen caracterizar por un elevado metabolismo de primer paso y una baja biodisponibilidad oral. Con la derivación portosistémica, la biodisponibilidad oral puede aumentar notablemente, así que las dosis orales deben reducirse en proporción a la cantidad de sangre derivada. Los cambios en la masa y la función hepáticas afectarán a los fármacos de capacidad limitada. En general, si la enfermedad hepática ha afectado negativamente a la concentración de albúmina sérica y al BUN, es probable que la capacidad metabólica intrínseca del hígado también se vea afectada negativamente. Sin embargo, si la unión a proteínas disminuye para un fármaco altamente unido a proteínas, de modo que una mayor parte del fármaco se libera, el aclaramiento hepático puede no verse afectado negativamente.
El aclaramiento renal se define como el volumen plasmático que se limpia totalmente del fármaco por unidad de tiempo (p. ej., L/minuto) durante su paso a través de los riñones. El aclaramiento renal de los fármacos depende principalmente del flujo sanguíneo renal; también se ve afectado por el pH de la orina, el grado de unión a proteínas plasmáticas, la capacidad de concentración de la orina y el uso concomitante de ciertos fármacos.
La concentración de creatinina sérica o el aclaramiento de creatinina sérica pueden usarse para valorar los cambios en el aclaramiento renal a medida que disminuye la función renal. Tanto la dosis como el intervalo pueden modificarse proporcionalmente. Para los medicamentos con una semivida corta, los intervalos se prolongan más adecuadamente (en comparación con la dosis decreciente) a medida que aumenta la concentración de creatinina sérica; para los medicamentos que se acumulan debido a una semivida prolongada, la dosis o el intervalo pueden reducirse o prolongarse de forma proporcional, respectivamente.
Constante de la tasa de eliminación
La semivida de eliminaciónes el tiempo transcurrido para que la Cp se reduzca al 50 %.
La semivida de eliminación se deriva de la constante de tasa de eliminación, kel, que es la pendiente del componente terminal de la curva de concentración plasmática frente al tiempo. Como parámetro híbrido, la kel se ve afectada tanto por el Cl como por el Vd. El Cl determina la disminución de la Cp; por tanto, cuanto mayor es el volumen de fármaco eliminado, más pronunciada es la pendiente, o la kel. El impacto del Vd sobre la semivida refleja su efecto sobre la Cp: un mayor Vd significa que hay menos fármaco en el volumen de sangre depurado por el hígado o los riñones. Por tanto, la tasa de eliminación disminuye a medida que el Vd aumenta, dando lugar a una relación inversa.
La semivida de eliminación se calcula como sigue:

donde ln 2 ≈ 0,693.
La relación entre kel y t1/2 refleja el hecho de que t1/2 se convierte en la pendiente de la curva cuando la concentración disminuye en un 50 % (es decir, C1/C2 = 2, donde C1 es la concentración en el tiempo 1 y C2 es la concentración en el tiempo 2). Dado que la t1/2 es inversamente proporcional a la kel, la t1/2 es directamente proporcional al Vd (un mayor Vd da lugar a una semivida más larga) e inversamente proporcional al Cl.
Obsérvese que el Cl y el Vd pueden alterarse profundamente, pero es posible que la t1/2 no cambie. Por ejemplo, en un animal deshidratado debido a una disfunción renal, el Cl puede disminuir en un 50 %, duplicando la t1/2. Sin embargo, si el animal está notablemente deshidratado, entonces el Vd disminuirá debido a la contracción del volumen de líquido extracelular. Dado que hay más fármaco en cada ml de sangre eliminada por el riñón, se puede eliminar la misma cantidad de fármaco y, como tal, la kel o la t1/2 pueden no cambiar.

La semivida de eliminación determina la cantidad de tiempo hasta el estado estacionario y la cantidad de tiempo que tarda un fármaco en eliminarse del organismo después de interrumpir su administración. Después de suspender un fármaco, el 50 % del fármaco se elimina en una semivida, el 75 % en la segunda semivida (la mitad del 50 %), el 87,5 % en la tercera y así sucesivamente. Con fines prácticos, la mayor parte del fármaco se elimina tras 3-5 semividas.
La semivida de eliminación, junto con las tolerancias, determina los tiempos de retirada o descarte de la leche en los animales de abasto y los tiempos de retirada para los animales que compiten en eventos deportivos regulados. La relación entre el intervalo de dosificación y la semivida de eliminación también determina si un fármaco fluctuará o se acumulará durante un intervalo de dosificación.
Curvas de concentración de dosis única tras la administración extravascular de fármacos
Cuando se administra un fármaco por vía extravascular, las concentraciones plasmáticas del fármaco aumentan hasta que se alcanza un pico o una concentración máxima del fármaco (Cmáx). Después de pasar a la circulación, el fármaco está sujeto de forma inmediata y simultáneamente a su distribución, biotransformación y excreción. La curva de concentración-tiempo después de la administración extravascular tiene una intersección y una pendiente en Y adicionales, y la pendiente refleja la constante de velocidad de absorción, ka. La semivida de absorción es el tiempo que transcurre a medida que el sistema absorbe el 50 % del fármaco. La absorción suele ser lo suficientemente lenta como para que la distribución del fármaco sea enmascarada por la fase de absorción. Por tanto, a medida que las concentraciones plasmáticas del fármaco disminuyen después de que se alcanza la Cmáx, la pendiente suele reflejar la kel.
El término biodisponibilidad se usa para expresar la tasa y el grado de absorción de un fármaco.
Concentración plasmática en estado estacionario (administración repetida o infusión continua IV) de fármacos
En algunos casos, con una sola dosis de un fármaco se obtiene el efecto terapéutico deseado. Sin embargo, con frecuencia es necesario mantener durante más tiempo las concentraciones del fármaco dentro del rango terapéutico para conseguir una respuesta satisfactoria. En lugar de administrar grandes dosis, que podrían dar como resultado concentraciones plasmáticas potencialmente tóxicas del fármaco, las múltiples dosis permiten intervalos regulares y más seguros. Para los fármacos con una semivida muy corta, el fármaco puede administrarse a través de un catéter como una infusión continua, que es esencialmente una administración IV continuada. La tasa de administración depende de la cantidad de fluctuación en la concentración del fármaco que puede producirse durante un intervalo de dosificación, que a su vez está determinada por la relación entre la t1/2 y el intervalo de dosificación, T.
Si un fármaco se administra a un intervalo sustancialmente más largo que su semivida, la mayor parte del fármaco se eliminará durante cada intervalo de dosificación. Por tanto, queda poco fármaco cuando se administra la dosis siguiente, y las concentraciones plasmáticas del fármaco fluctuarán (desde la concentración máxima del fármaco [Cmáx] hasta la concentración mínima del fármaco [Cmín]) durante el intervalo de dosificación.
Por ejemplo, si un fármaco con una semivida de 4 horas se administra cada 12 horas, el 87,5 % del fármaco se eliminará durante cada intervalo de dosificación. Con cada dosis, existe el riesgo de que las concentraciones del fármaco se vuelvan subterapéuticas; el aumento de la dosis dará lugar a un pequeño aumento de la Cmín, pero puede incrementar sustancialmente la Cmáx, aumentando así el riesgo de toxicidad.
Una respuesta más apropiada sería disminuir el intervalo de dosis. Sin embargo, esto puede ser necesario solo si la eficacia del fármaco depende de su presencia. Por ejemplo, esta magnitud de fluctuación puede ser aceptable para un antimicrobiano dependiente de la concentración como la gentamicina. Sin embargo, si el fármaco es un anticonvulsivo, el riesgo de convulsiones aumenta justo antes de la siguiente dosis. Si el fármaco depende del tiempo, las concentraciones del fármaco pueden descender por debajo de la concentración inhibitoria mínima del microbio infectante.
A diferencia de los fármacos con una semivida corta, los fármacos con una semivida larga en comparación con el intervalo de dosificación se acumularán con cada dosis, porque gran parte del fármaco permanece en el organismo cuando se administra la siguiente dosis. Los fármacos con una larga semivida comienzan a acumularse con la primera dosis y continuarán haciéndolo hasta que se alcance un equilibrio de estado estacionario tal que la cantidad de fármaco eliminada durante cada intervalo de dosificación sea equivalente a la cantidad de fármaco administrada durante ese mismo intervalo. La tasa de acumulación describe la magnitud del incremento de la Cmáx o la Cmín en estado estacionario en comparación con la primera dosis. Cuanto mayor es la semivida en comparación con el intervalo de dosificación, mayor es la tasa de acumulación.
Al igual que con la eliminación del fármaco, para fines prácticos, el estado estacionario se alcanza en 3-5 semividas, independientemente del fármaco o la dosis, siempre que la preparación y la pauta posológica sean las mismas. En tales casos, el 50 % de la meseta o la concentración en estado estacionario se alcanzará en una t1/2, el 75 % a las dos t1/2, el 87,6 % a las tres t1/2 y el 93,6 % a las cuatro t1/2. La respuesta al fármaco, ya sea por su eficacia o por su toxicidad, no puede evaluarse hasta que se alcanza el estado estacionario. Debido a que la cantidad de fármaco en el organismo es grande en comparación con cada dosis, es difícil manipular las concentraciones plasmáticas del fármaco para tales fármacos, porque los cambios requieren una dosificación de tres a cinco semividas con la nueva dosis.
Si el tiempo para alcanzar el estado estacionario, y por tanto el tiempo para el efecto terapéutico, es inaceptable, las concentraciones plasmáticas del fármaco en el estado estacionario pueden alcanzarse más rápidamente mediante la administración de una o varias dosis de carga, como sigue:

donde D es la dosis (en mg/kg), Vd es el volumen de distribución en estado estacionario (en ml/kg) y Cp es la concentración plasmática objetivo (en mg/ml). Si el fármaco se administra PO, la dosis debe tener en cuenta la biodisponibilidad:

donde F es la biodisponibilidad (en %). Sin embargo, el fármaco no se encontrará en estado estacionario, sino solo en concentraciones estables.
Si la dosis de mantenimiento no mantiene la dosis de carga conseguida, cuando se alcanza el estado estacionario a la dosis de mantenimiento, las concentraciones plasmáticas del fármaco pueden aumentar para causar intoxicación o disminuir hasta una concentración subterapéutica.
Los fármacos con una semivida muy corta se suelen administrar a un ritmo de infusión continua en animales en estado crítico. En estos casos, el intervalo es infinitamente corto en comparación con la semivida, y el fármaco se acumula hasta alcanzar el estado estacionario. La velocidad de infusión se calcula como sigue:

donde I es la tasa de infusión (en μg/kg/minuto), Cl es el aclaramiento (en ml/kg /minuto) y Cp es la concentración plasmática objetivo (en μg/ml).
Se debe administrar una dosis de carga si el tiempo para alcanzar el estado estacionario es inaceptablemente largo.



{kind=link}