




La farmacodinámica es la rama de la farmacología dedicada a los efectos bioquímicos y fisiológicos de los fármacos y de sus mecanismos de acción en el organismo o en microorganismos y otros parásitos dentro del organismo o sobre él. Se consideran tanto la acción del fármaco, que se refiere a la fase inicial consecuencia de la interacción fármaco-receptor, como el efecto del fármaco, que se refiere a los efectos subsiguientes.
Por el contrario, la farmacocinética es la rama de la farmacología que se centra en el curso temporal de las concentraciones del fármaco. Sin embargo, la farmacocinética y la farmacodinámica están interrelacionadas en el sentido de que las concentraciones del fármaco impulsan los efectos clínicos.
Los mecanismos de acción de los fármacos pueden agruparse ampliamente como mediados por proteínas o no mediados por proteínas:
Mecanismos de los fármacos mediados por proteínas
Los fármacos o tóxicos se dirigen con mayor frecuencia a proteínas específicas, como enzimas, transportadores y canales de iones, proteínas receptoras o ADN, o estructuras subcelulares como los microtúbulos. Algunos ejemplos de clases de fármacos (y sus dianas proteicas) incluyen los siguientes:
AINE (ciclooxigenasa).
Glucósidos digitálicos (Na+/ K+-ATPasa).
Anestésicos locales (canales de sodio).
Inhibidores de la bomba de protones (H+/K+-ATPasa de las células parietales gástricas).
Diuréticos del asa (Na+/ K+/ 2Cl−-simportador en la rama ascendente gruesa del asa renal de Henle).
Fluoroquinolonas (ADN girasa bacteriana).
Los fármacos alcaloides de la vinca como la vincristina y la vinblastina interactúan y alteran los microtúbulos del huso mitótico.
Mecanismos de fármacos no mediados por proteínas
Aunque la mayoría de los fármacos interactúan con proteínas diana (receptores y enzimas), los fármacos también pueden ejercer un efecto a través de interacciones físicas con los líquidos o tejidos orgánicos. Algunos ejemplos de estos fármacos incluyen el diurético osmótico manitol y los antiácidos administrados por vía oral, que cambian la osmolalidad o el pH de los líquidos corporales y no interactúan directamente con tejidos u órganos.
Mecanismos de transducción de señales mediados por receptores en animales
La regulación de los receptores celulares y los mecanismos de transducción de señales son importantes para el impacto de los fármacos en los pacientes. La mayoría de los fármacos actúan a través de receptores conectados a mecanismos de transducción de señales.
Las características clave de los receptores incluyen lo siguiente:
Especificidad estructural relativa para una clase de compuesto.
Número finito (es decir, los receptores son saturables).
Relación con un efecto biológico distinto.
La especificidad del receptor no es absoluta, sino que depende de la afinidad de unión entre un ligando (es decir, un fármaco) y su sitio de unión en un receptor. A medida que aumenta la dosis y la concentración de un fármaco, puede aumentar su interacción con receptores no diana, dando lugar a efectos clínicos adversos.
La transducción de señales se suele lograr con uno o más procesos celulares clave, cada uno de los cuales permite que una señal (p. ej., un fármaco) transmita su efecto desde el exterior de la célula al compartimento intracelular. Hay varias categorías de sistemas de transducción de señales (Tipos generales de transducción de señales mediada por receptores).
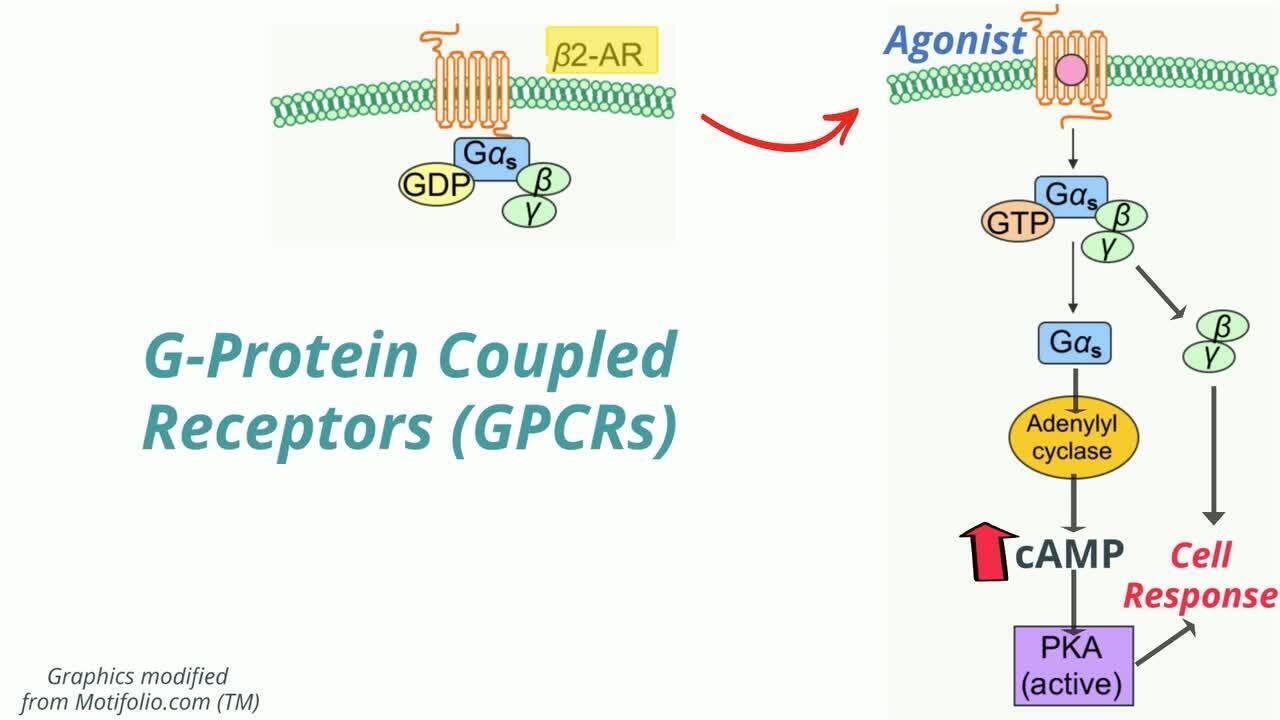

Los receptores transmembrana se clasifican como ionotrópicos (ligados a canales iónicos) o metabotrópicos (ligados a procesos bioquímicos). Los receptores ionotrópicos son canales iónicos transmembrana activados por ligando. Los ejemplos incluyen los canales de cloro ligados al ácido gamma-aminobutírico (GABA) y los receptores nicotínicos de acetilcolina ligados a los canales de sodio.


Por cortesía del Dr. Duncan C. Ferguson.
Los receptores metabotrópicos actúan directa o indirectamente sobre las enzimas de transducción de señales o están unidos a enzimas que tienen un dominio extracelular que reconoce un fármaco y un dominio intracelular que cataliza una respuesta bioquímica. Este subtipo de receptor incluye los receptores acoplados a la proteína G (GPCR).
Otros receptores son los ligados a la guanilil ciclasa (GC). En este caso, los receptores de la membrana plasmática tienen actividad guanilato ciclasa y el GMP cíclico formado activa la proteína cinasa G (PKG), que fosforila proteínas asociadas con la acción celular. Algunos ejemplos de este tipo de receptor son los receptores para el péptido natriurético auricular (PNA) y el óxido nítrico (directamente o vía receptores muscarínicos).
Otros receptores son los relacionados con las enzimas cinasas, como el receptor de la insulina, que tiene actividad enzimática intrínseca de tirosina cinasa y puede transferir un grupo fosfato del ATP a una proteína (en este caso, el sustrato del receptor de insulina 1 [IRS-1]) en una célula. Esto activa proteínas adicionales a través de la fosforilación, lo que da lugar a la translocación de la glucosa por el transportador de glucosa tipo 4 (GLUT4). Los receptores de este tipo pueden regularse de diversas formas mediante la fosforilación y la desfosforilación.
Finalmente, hay receptores intracelulares, incluidos los receptores citosólicos y los receptores nucleares. Algunos ejemplos de receptores nucleares incluyen los de las hormonas lipofílicas, como las hormonas tiroideas y esteroideas, que se unen a un elemento de respuesta del ADN, lo que provoca la inhibición o activación del ARNm y, finalmente, la transcripción de proteínas.
Unión fármaco-receptor en animales
La farmacocinética predice que las concentraciones plasmáticas (y tisulares) son proporcionales a la dosis administrada. Asimismo, la cantidad de fármaco unido al receptor también suele ser proporcional a la tasa de transducción de la señal (es decir, el efecto). Sin embargo, el paso intermedio de la unión al receptor añade un componente no lineal de modo que, en general, la respuesta o efecto farmacológico no está relacionada de forma lineal con la concentración y la dosis del fármaco.
La mayoría de los fármacos interactúan con los receptores en un equilibrio dinámico, lo que significa que existe una unión reversible al receptor. Debido a esta reversibilidad, la concentración plasmática impulsa la respuesta al fármaco de manera proporcional hasta que se alcanza el efecto máximo, a menudo, pero no siempre, cuando todos los receptores están ocupados. (Equilibrio dinámico de la interacción fármaco-receptor).


Por cortesía del Dr. Duncan C. Ferguson.


Por cortesía del Dr. Duncan C. Ferguson.


Por cortesía del Dr. Duncan C. Ferguson.
Sin embargo, la acción masiva de la unión predice una curva dosis-respuesta no lineal, que es hiperbólica pero que a menudo se muestra como un gráfico sigmoideo de la curva logarítmica (concentración) frente a la respuesta (Curva dosis-respuesta de unión fármaco-receptor). La constante de equilibrio de disociación (KD) define la mitad del rango operativo de concentración para la interacción fármaco-receptor, lo que significa que es la concentración a la que están ocupados el 50 % de los receptores. En general, un fármaco mostrará su rango completo de efecto dentro de un rango de concentración de 100 veces. Esto se ejemplifica en Curva dosis-respuesta de unión fármaco-receptor: el umbral de actividad del fármaco se produce aproximadamente a una concentración de 1 y se acerca a la unión máxima a una concentración de 100, con un 50 % del máximo alcanzado a una concentración de 10. La capacidad máxima de unión (Bmax) es representativa de la cantidad finita de proteína receptora presente en una célula o fracción subcelular.
Relación dosis-respuesta en animales
Dado que la concentración del fármaco está relacionada linealmente con la dosis, la relación entre la concentración del fármaco y el efecto puede describirse con una ecuación similar. (Curvas logarítmicas de concentración frente a respuesta).
Una comparación con la curva definida por la unión del fármaco muestra que el efecto del fármaco se describe mediante una ecuación similar en la que la unión máxima (Bmax) es un término análogo al efecto máximo (Emax) y la constante de equilibrio de disociación. KD se reemplaza por la concentración efectiva media (CE50).
Agonistas de receptores en animales
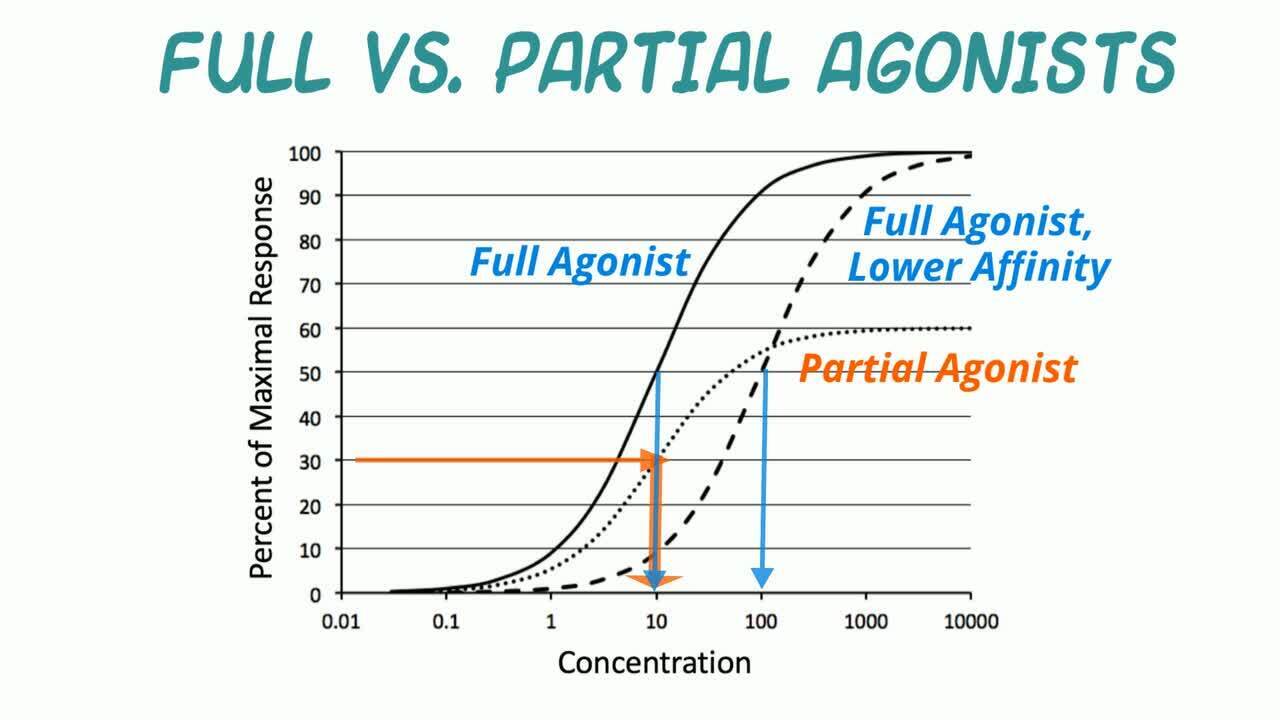
Los fármacos que impulsan una vía de señalización hacia delante, dando lugar a un efecto biológico a menudo asociado con un efecto terapéutico, se denominan agonistas.
Los compuestos que interactúan con los receptores se dividen en distintos tipos farmacológicos basados en su eficacia intrínseca en un sistema de transducción de señal de receptor. Un agonista completo logra el mayor efecto biológico en su concentración máxima eficaz. Un agonista parcial es un agonista para el cual el Emax, y por tanto la eficacia intrínseca, es menor que la de un agonista completo. (Curvas logarítmicas de concentración frente a respuesta).


Por cortesía del Dr. Duncan C. Ferguson.
Un ejemplo clásico de un agonista parcial es la buprenorfina, un agonista parcial opiáceo mu. Su actividad se compara con la de la morfina, un agonista mu completo. Sin embargo, la buprenorfina es aproximadamente 30 veces más potente que la morfina porque se une al receptor con mayor afinidad. El beneficio clínico de un agonista parcial estaría asociado con la seguridad del techo por su efecto (es decir, su Emax). Un Emax más bajo se asociaría con una menor tendencia al efecto adverso de depresión respiratoria con los fármacos opiáceos.
Comprensión de la transducción de receptores de fármacos en animales
Cuando dos fármacos se unen al mismo receptor conectado al mismo sistema de transducción de señales, pero conducen a diferentes efectos máximos, tienen una eficacia intrínseca diferente. Un receptor puede estar en dos estados: el estado de reposo (R) y el estado activado (R*). Estos estados están en equilibrio entre sí, pero pueden verse afectados por el tipo de fármaco que se une a ellos.
Cuando un agonista completo interactúa con el receptor, desplaza el equilibrio hacia la forma activa del receptor y tiene una afinidad preferencial por la forma activada en relación con la forma en reposo. Cuanto mayor sea la selectividad conformacional para R*, mayor será la eficacia. Con un agonista parcial, el efecto máximo es menor (es decir, parcial) que el de un agonista puro. Considerando la teoría del receptor, el agonista parcial, aunque favorece a R*, no desplaza el equilibrio conformacional hacia R* tan eficazmente como un agonista puro. Por eso, incluso con el efecto máximo, el efecto es menor que el efecto máximo para un agonista completo.
Una analogía útil es comparar el efecto de un fármaco con el de una bicicleta impulsada por un pedal y una cadena con múltiples marchas. En esta analogía, el mismo número de rotaciones de los pedales (es decir, concentración y afinidad por el receptor) de un agonista completo movería la bicicleta más abajo en la carretera que un agonista parcial (es decir, el Emax para un agonista completo es mayor que el Emax para un agonista parcial). Para completar la analogía, una marcha baja se relacionaría con un agonista de baja afinidad y una marcha más alta se relacionaría con un agonista de alta afinidad, afectando principalmente a la velocidad alcanzada por la bicicleta (efecto) a una determinada tasa de pedaleo (concentración), y no al límite de la distancia recorrida (Emax).
Aunque puede parecer contradictorio utilizar un fármaco que puede tener solo una parte del efecto máximo, esto puede ser deseable en el caso de algunos fármacos, como los agonistas opiáceos. La morfina y la oximorfona, por ejemplo, se consideran agonistas puros o completos. Sin embargo, sus efectos beneficiosos dependientes de la dosis y sus efectos adversos (p. ej., supresión del centro respiratorio, que puede conducir a la muerte) se superponen considerablemente, lo que hace que su administración sea bastante peligrosa. Sin embargo, los agonistas opiáceos parciales como el butorfanol o la buprenorfina pueden administrarse de forma más segura porque sus efectos beneficiosos se estabilizan antes de alcanzar el punto en el que suele observarse el efecto adverso de la depresión respiratoria.
Antagonistas en animales
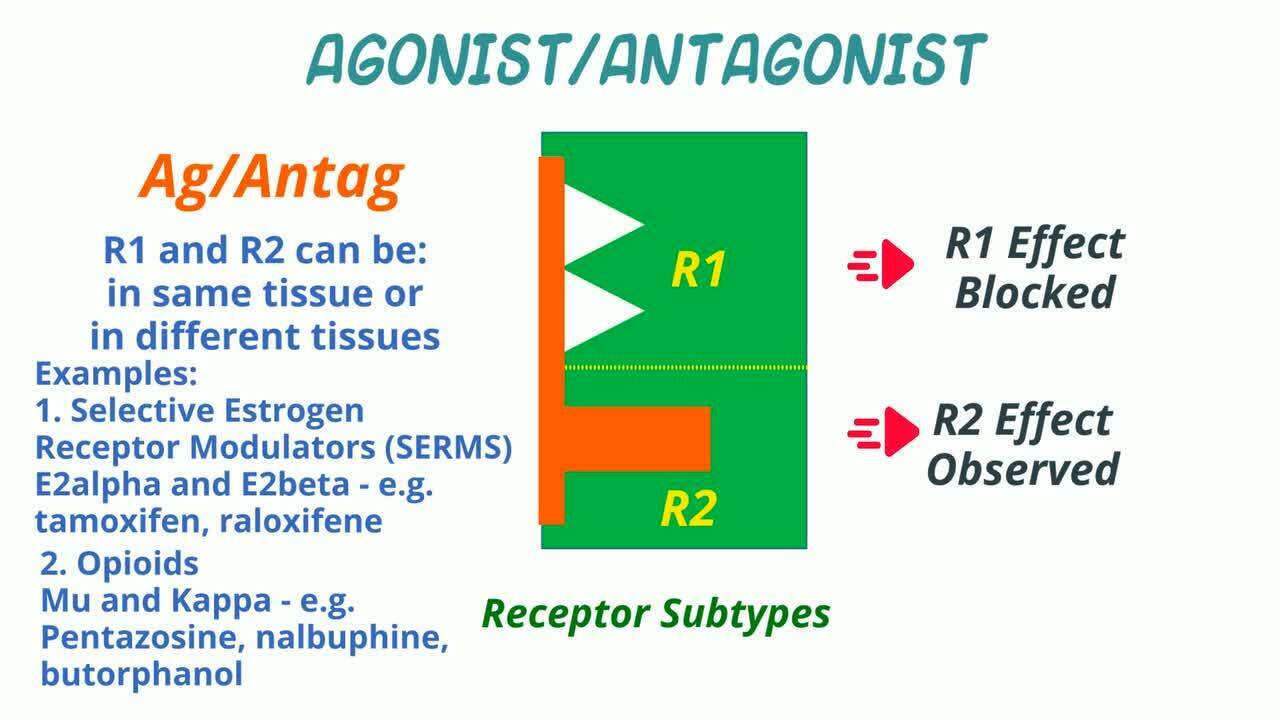
Los fármacos que tienden a actuar sobre los receptores para disminuir ciertas vías de señalización se conocen como antagonistas, agonistas/antagonistas y agonistas inversos. Los fármacos antagonistas de los receptores bloquean la acción de un compuesto endógeno o exógeno, ya sea por competencia directa en el receptor o por alteración de la función del receptor.
La mayoría de los fármacos antagonistas del receptor se consideran antagonistas competitivos, lo que significa que compiten con un agonista por unirse a un receptor por acción masiva, pero no alteran el receptor. Desde una perspectiva clínica, los antagonistas puros no tienen una eficacia intrínseca propia, pero compiten con el agonista en función de su afinidad por el receptor (por lo general diseñado para ser mayor). Sin embargo, su efecto solo se produce bloqueando el efecto de un agonista endógeno o exógeno (puro o parcial).
Otros términos usados para antagonistas son "bloqueantes" y fármacos "líticos". Por ejemplo, el atenolol, un antagonista de los receptores beta1, a menudo se denomina betabloqueante y se considera un fármaco simpaticolítico.


Por cortesía del Dr. Duncan C. Ferguson.
Una gráfica logarítmica de concentración frente a efecto ( See figure Dosis de antifúngicos para tratamiento respiratorio) muestra que la adición de un antagonista competitivo desplaza la curva dosis-respuesta de un agonista hacia la derecha, lo que significa que la CE50 será aparentemente más alta en presencia de un antagonista competitivo. Aunque no todos los antagonistas competitivos tienen esta alta afinidad, cuando la tienen y las concentraciones de antagonista se mantienen, casi parecen irreversibles porque los agonistas disponibles no pueden administrarse en cantidades adecuadas para competir con el antagonista. Y lo que es más importante para el veterinario, el efecto que se produce en la CE50 original es minúsculo, lo que provoca el efecto clínico del bloqueo.
Conceptualizando esto con la teoría del receptor, un antagonista competitivo es un compuesto que se une con igual afinidad a los receptores en su estado de reposo (R) y estado activado (R*). Competirá con un agonista total o parcial por la unión al receptor, pero no modificará el equilibrio conformacional y, por tanto, no producirá ningún efecto por sí solo.
En la analogía del engranaje de la bicicleta, el bloqueo por un antagonista competitivo sería similar a una situación en la que la cadena se ha desprendido del plato dentado. Los intentos de pedalear la bicicleta no producen ningún movimiento hacia delante (es decir, debido a la presencia del antagonista competitivo, una concentración normalmente eficaz de un agonista ya no produce un efecto biológico).
Antagonistas no competitivos en animales
Un pequeño número de fármacos se considera antagonistas no competitivos. Estos fármacos pueden producir una modificación covalente del receptor, eliminando su capacidad para unirse al agonista, o pueden interactuar con un sitio alostérico cerca del receptor, cambiando la configuración del receptor. De cualquier forma, los antagonistas no competitivos disminuyen el Emax y ninguna concentración de agonista es capaz de superar este efecto. (Efecto del antagonista no competitivo sobre la relación dosis-respuesta).


Por cortesía del Dr. Duncan C. Ferguson.
Un ejemplo de este tipo de fármaco utilizado en medicina veterinaria es el antagonista de los receptores alfa adrenérgicos fenoxibenzamina, que se utiliza con mayor frecuencia para los espasmos urinarios poscateterización. Este fármaco interactúa con el receptor y después de un periodo de tiempo se une de forma covalente a él. La relevancia clínica es que este fármaco necesita algún tiempo para alcanzar su máxima acción. Sin embargo, el efecto máximo que puede alcanzar un agonista está disminuido, al igual que con una disminución del número de receptores (disminución de la Bmax). Solo la síntesis de nuevos receptores puede devolver el efecto máximo a los niveles previos al antagonista.
Agonistas/antagonistas en animales
Un agonista/antagonista es un fármaco que es un agonista en algunas condiciones y un antagonista en otras (otro receptor o subtipo de receptor u otro tejido). En su papel de antagonista puede bloquear la actividad de otros agonistas. Cuando actúa de forma diferente según el tejido, puede denominarse modulador selectivo del receptor. Los moduladores selectivos del receptor de estrógenos (SERM) como el tamoxifeno o el raloxifeno son algunos ejemplos.
En medicina veterinaria, un ejemplo destacado de agonistas-antagonistas son los opiáceos. Los fármacos que actúan como agonistas del receptor opiáceo kappa y antagonistas del receptor opiáceo mu incluyen la pentazocina y la nalbufina. El butorfanol es también un agonista puro del receptor opiáceo kappa, un agonista parcial del receptor opiáceo mu y un antagonista del receptor opiáceo delta.
Agonistas inversos en animales
Los fármacos también pueden contrarrestar una vía de transducción de señales interactuando directamente con el sistema de transducción. Hay receptores que están asociados con la transducción intrínseca incluso cuando no hay ocupación del receptor. Los agonistas inversos interactúan con la vía de transducción de señales, pero disminuyen la actividad intrínseca de la vía. (Curva dosis-respuesta para los agonistas inversos en comparación con los agonistas completos).
Conceptualizando esto con la teoría del receptor, un agonista inverso se une con mayor afinidad a los receptores en su estado de reposo (R) que a los receptores en su estado activado (R*), desplazando el equilibrio conformacional hacia R, la forma de receptor que es menos eficaz para impulsar las vías de transducción de la señal hacia delante. Cuando interactúa con un sistema donde el receptor no ligado tiene actividad constitutiva, un agonista inverso sirve para disminuir el efecto por debajo de los niveles constitutivos (es decir, la curva dosis-respuesta es la inversa de la de un agonista total o parcial).


Por cortesía del Dr. Duncan C. Ferguson.
En la analogía de la bicicleta y el engranaje, el agonista inverso haría que la bicicleta fuera en la dirección opuesta a un agonista puro. También podría imaginarse como si la bicicleta fuera cuesta abajo (actividad libre de agonistas intrínsecos) y pudiera ralentizarse frenando (actividad reducida en presencia de un agonista inverso).
Fenómenos del receptor de relevancia clínica en animales
La respuesta a un determinado fármaco puede variar entre pacientes. Algunos pacientes responden de manera constante más a un fármaco y otros menos. También pueden producirse cambios en la respuesta al fármaco a lo largo del tiempo en el mismo paciente porque la cantidad del receptor del fármaco, la afinidad y la actividad de transducción de señales varían con el tiempo.
Hiporreactividad o tolerancia
El organismo tiene múltiples mecanismos mediante los cuales puede adaptarse a la administración de múltiples dosis de un fármaco o tóxico, y tanto el receptor como los mecanismos de transducción de señales pueden estar implicados en esta adaptación. El término general para la hiporreactividad desarrollada es tolerancia.
Los mecanismos que provocan la tolerancia después de la administración crónica pueden ser difíciles de distinguir en el paciente individual a menos que se realice un estudio completo de respuesta a la dosis.
Pérdida rápida del efecto farmacológico: taquifilaxia
La taquifilaxia se refiere a la pérdida de efecto relativamente rápida en un periodo corto de tiempo (de minutos a días). Por ejemplo, los aerosoles nasales descongestionantes y las gotas para los ojos a menudo incluyen agonistas de los receptores alfa adrenérgicos (p. ej., fenilefrina) para la descongestión tópica por vasoconstricción de los vasos sanguíneos de la mucosa nasal. El uso prolongado (más de varios días) de un descongestionante nasal puede provocar una disminución del efecto con el tiempo. El mecanismo de esta taquifilaxia es la regulación a la baja (reducción) mediada por los receptores alfa adrenérgicos y la desensibilización de la respuesta inducida por fármacos (véase más adelante).
Pérdida del efecto farmacológico: desensibilización y regulación a la baja de los receptores y agotamiento de los mediadores
La respuesta de un fármaco que estimula los receptores acoplados a proteína G puede disminuir rápidamente o más lentamente. Ejemplificado por el receptor beta adrenérgico, la desensibilización se suele asociar con la fosforilación del receptor. En un estado desensibilizado, la curva dosis-efecto se desplazaría en paralelo a concentraciones más altas (como se observa con los antagonistas competitivos en Efecto del antagonista competitivo sobre la relación dosis-respuesta y Curva dosis-respuesta para los agonistas inversos en comparación con los agonistas completos), y la CE50 aparente sería mayor, lo que significa que la misma dosis (concentración) del fármaco provocaría un efecto menor que inicialmente. Cuando se elimina el agonista, las fosfatasas celulares invierten el efecto.
El segundo mecanismo por el cual una célula o tejido puede modular la acción de un fármaco es la regulación a la baja, la reducción del número de receptores. En una gráfica logarítmica de concentración frente a efecto, la pérdida de receptores aparece como se muestra en Efecto del antagonista no competitivo sobre la relación dosis-respuesta. Con algunos receptores acoplados a proteína G, incluidos los receptores beta adrenérgicos, la estimulación crónica provoca un aumento de la fosforilación del receptor, que después lo transmite a los lisosomas que destruyen el receptor fosforilado, reduciendo eficazmente el número de receptores disponibles en la membrana plasmática. Esta regulación a la baja se caracteriza por un Emax más bajo, un efecto secundario de la disminución del número de receptores (Bmax).
Un tercer mecanismo para la reducción de los efectos de un fármaco es el agotamiento de mediadores de transducción de señales. Por ejemplo, el agente adrenérgico anfetamina da lugar a la liberación de catecolaminas sinápticas. La administración crónica puede dar lugar al agotamiento de las reservas de catecolaminas.
Hiperreactividad
Puede producirse un aumento de la reacción a un fármaco cuando hay una reducción de los ligandos fisiológicos endógenos del receptor, que puede producir una supersensibilización (asociada con una disminución de la EC50 o un aumento del Emax). Por ejemplo, los antagonistas beta adrenérgicos como el propranolol o el atenolol, utilizados para controlar la frecuencia cardiaca y la presión arterial, disminuyen la señalización a través de la adenilato ciclasa. La reducción del AMPc y la fosforilación finalmente dan como resultado el reclutamiento y la regulación positiva de los receptores beta en la membrana celular. La retirada de estos fármacos se asociaría inicialmente con una hiperreactividad a los mediadores endógenos (p. ej., adrenalina), lo que podría conducir a una hipertensión de rebote. El efecto clínico se conoce como sobreimpulso y es la razón por la que se recomienda la retirada gradual de los fármacos autónomos siempre que sea posible para permitir que la respuesta celular y tisular vuelva a la normalidad.
Receptores de reserva
La capacidad de reserva del receptor proporciona un mecanismo para obtener la respuesta máxima a una concentración muy baja de agonista a pesar de una afinidad relativamente baja (es decir, alta KD) por el receptor.


Por cortesía del Dr. Duncan C. Ferguson.
Con algunos fármacos, el efecto biológico máximo se puede conseguir a dosis o concentraciones mucho más bajas de lo esperado, según la afinidad de unión del receptor. En lugar de un cambio en un paciente, este fenómeno es una característica incorporada del sistema receptor; sin embargo, puede variar considerablemente entre especies (consúltese la figura "Ejemplo de receptores de reserva").
Un ejemplo clásico de receptores de reserva se encuentra en el sistema de receptores nicotínicos de la acetilcolina. Estos receptores de baja afinidad están ligados a los canales iónicos de sodio. La baja afinidad de estos receptores permite que la acetilcolina sináptica (ACh), por lo general a una concentración elevada de 1 mmol/L, se disocie rápidamente de su receptor para terminar con el estímulo neurotransmisor iniciador. La ACh se destruye después por la acetilcolinesterasa sináptica. Una vez despolarizados, estos receptores sufren una desensibilización temporal. Como resultado, para mantener el potencial de estimulación repetitiva a ritmos fisiológicos normales, se deben reclutar receptores adicionales para mantener la contracción.
Un escenario clínico que ilustra la importancia de los receptores de reserva es la administración de d-tubocurarina, que bloquea competitivamente los receptores nicotínicos en la membrana plasmática postsináptica del músculo esquelético y se usa durante los protocolos anestésicos para optimizar la relajación muscular. Como los animales suelen estar intubados por vía endotraqueal durante el bloqueo neuromuscular, es importante una duración de acción predecible y razonable. Para el efecto de relajación del músculo esquelético, el antagonista competitivo d-tubocurarina debe bloquear no solo los receptores activos, sino también los receptores de reserva.
Puntos clave
La mayoría de los fármacos actúan mediante la interacción con proteínas celulares: enzimas, transportadores o receptores.
Los efectos farmacodinámicos de las interacciones fármaco-receptor suelen ser de naturaleza reversible y se definen por la concentración del fármaco y la constante de unión en equilibrio.
La relación de unión fármaco-receptor puede representarse como una curva logarítmica dosis-respuesta sigmoidea.
Los fármacos que actúan sobre los receptores para impulsar una vía de señalización son agonistas totales o parciales y los que actúan sobre los receptores para disminuir las vías de señalización son antagonistas, agonistas/antagonistas o agonistas inversos.
La respuesta a un determinado fármaco puede variar entre pacientes, según la cantidad del receptor del fármaco, la afinidad y la actividad de transducción de señales.
Para más información
Ferguson, DC. Principles of Pharmacodynamics and Toxicodynamics. En Haschek and Rousseaux's Handbook of Toxicologic Pathology. Elsevier Inc.; 2013: 61-76. https://doi.org/10.1016/B978-0-12-415759-0.00003-0
Consulte también la información para propietarios sobre fármacos y vacunas.


